GEO生信数据挖掘(三)芯片探针ID与基因名映射处理
最佳答案 问答题库878位专家为你答疑解惑
检索到目标数据集后,开始数据挖掘,本文以阿尔兹海默症数据集GSE1297为例
目录
处理一个探针对应多个基因
1.删除该行
2.保留分割符号前面的第一个基因
处理多个探针对应一个基因
详细代码案例一删除法
详细代码案例二 多个基因名时保留第一个基因名
小结
更新版本的代码全文
上节我们下载了基因芯片平台文件并注释,我们发现存在一个芯片探针ID匹配到多个基因的情况,本节来介绍处理方案。
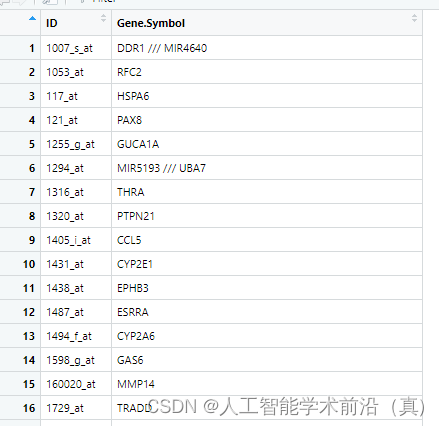
处理一个探针对应多个基因
我们通过简单检索发现两种方法:1.删除操作 2.保留分割符号前面的第一个基因
1.删除该行
#处理一个探针对应多个基因
#方案一:【删除该行】explan_final <- data.frame(explan_final[-grep("///",explan_final$"Gene.Symbol"),]) #去一对多,grep是包含的意思,-就是不包含
2.保留分割符号前面的第一个基因
#方案二:【保留第一个基因名】
ids = platform_file_set #探针列名和基因名两列
library(tidyverse)
test_function <- apply(ids,1,function(x){paste(x[1],str_split(x[2],'///',simplify=T),sep = "...")})
x = tibble(unlist(test_function))colnames(x) <- "ttt"
ids <- separate(x,ttt,c("ID","Gene.Symbol"),sep = "\\...")
dim(ids) #探针列名和基因名两列显然,第一个发现非常简单,在使用merge函数匹配时,会剔除更多的基因。第二个方法,会保留更多基因。
处理多个探针对应一个基因
表达矩阵中还有一个问题,如下图所示,很多探针指向同一个基因。
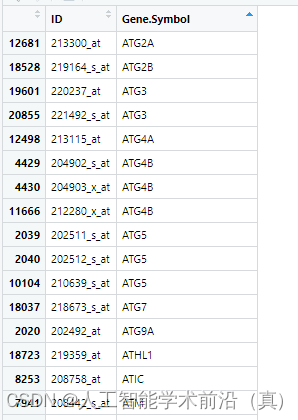
#把重复的Symbol 取每个基因所有探针的平均值或最大值作为基因的表达量
matrix <- aggregate(.~Gene.Symbol, matrix, mean) ##把重复的Symbol取平均值
matrix <- aggregate(.~Gene.Symbol, matrix, max) ##把重复的Symbol取最大值
详细代码案例一删除法
# 安装并加载GEOquery包 library(GEOquery)# 指定GEO数据集的ID gse_id <- "GSE1297"# 使用getGEO函数获取数据集的基础信息 gse_info <- getGEO(gse_id, destdir = ".", AnnotGPL = F ,getGPL = F) # Failed to download ./GPL96.soft.gz!# 提取基因表达矩阵 expression_data <- exprs(gse_info[[1]])#查看平台文件列名 colnames(annotation)#打印项目文件列表 dir() # 读取芯片平台文件txt platform_file <- read.delim("GPL96-57554.txt", header = TRUE, sep = "\t", comment.char = "#")#查看平台文件列名 colnames(platform_file)# 假设芯片平台文件中有两列,一列是探针ID,一列是基因名 #probe_names <- platform_file$ID #gene_symbols <- platform_file$Gene.Symbol platform_file_set=platform_file[,c(1,11)]#将Matrix格式表达矩阵转换为data.frame格式 exprSet <- data.frame(expression_data)#给表达矩阵新增加一列ID exprSet$ID <- rownames(exprSet) # 得到表达矩阵,行名为ID,需要转换,新增一列#矩阵表达文件和平台文件有相同列‘ID’,使用merge函数合并 express <- merge(x = exprSet, y = platform_file_set, by.x = "ID")#删除探针ID列 express$ID =NULLdim(express) exprSet = express #查看多少个基因重复了 table(duplicated(exprSet$Gene.Symbol))#处理重复基因,计算行平均值方案1 #rowMeans = apply(exprSet[,c(1:12)],1,function(x) mean(as.numeric(x), na.rm = T))####计算行平均值#处理重复基因,计算行平均值方案2 #matrix <- aggregate(.~Gene.Symbol, matrix, mean) ##把重复的Symbol取平均值 #row.names(matrix) <- matrix$Gene.Symbol #把行名命名为SYMBOL#处理重复基因,计算行平均值方案3 library(limma) #avereps 函数 exp_unique<-avereps(exp_symbol[,-c(32,ncol(exp_symbol))],ID=exp_symbol$Gene.Symbol)##把重复的Symbol取平均值#排序 exprSet = exprSet[order(rowMeans, decreasing = T),] dim(exprSet)#去掉重复基因 exprSet_2 = exprSet[!duplicated(exprSet[, dim(exprSet)[2]]),] dim(exprSet_2)#去掉缺失值 exprSet_na = na.omit(exprSet_2) explan_final = exprSet_na[exprSet_na$Gene.Symbol != "",] dim(explan_final)#处理一个探针对应多个基因[删除法] explan_final <- data.frame(explan_final[-grep("///",explan_final$"Gene.Symbol"),]) #去一对多,grep是包含的意思,-就是不包含 dim(explan_final)rownames(explan_final) <- explan_final$Gene.Symbol dim(explan_final) explan_final <- explan_final[,c(1:31)] # 此时explan_final为所需文件,行为基因,列为样本
> dim(explan_final)
[1] 12548 31
详细代码案例二 多个基因名时保留第一个基因名
# 安装并加载GEOquery包
library(GEOquery)# 指定GEO数据集的ID
gse_id <- "GSE1297"# 使用getGEO函数获取数据集的基础信息
gse_info <- getGEO(gse_id, destdir = ".", AnnotGPL = F ,getGPL = F) # Failed to download ./GPL96.soft.gz!# 提取基因表达矩阵
expression_data <- exprs(gse_info[[1]])# 提取注释信息
annotation <- featureData(gse_info[[1]])#查看平台文件列名
colnames(annotation)#打印项目文件列表
dir() # 读取芯片平台文件txt
platform_file <- read.delim("GPL96-57554.txt", header = TRUE, sep = "\t", comment.char = "#")#查看平台文件列名
colnames(platform_file)# 假设芯片平台文件中有两列,一列是探针ID,一列是基因名
#probe_names <- platform_file$ID
#gene_symbols <- platform_file$Gene.Symbol
platform_file_set=platform_file[,c(1,11)]#一个探针对应多个基因名,保留第一个基因名
ids = platform_file_set
library(tidyverse)
test_function <- apply(ids,1,function(x){paste(x[1],str_split(x[2],'///',simplify=T),sep = "...")})
x = tibble(unlist(test_function))colnames(x) <- "ttt"
ids <- separate(x,ttt,c("ID","Gene.Symbol"),sep = "\\...")
dim(ids)#将Matrix格式表达矩阵转换为data.frame格式
exprSet <- data.frame(expression_data)
dim(exprSet)#给表达矩阵新增加一列ID
exprSet$ID <- rownames(exprSet) # 得到表达矩阵,行名为ID,需要转换,新增一列
dim(exprSet)
#矩阵表达文件和平台文件有相同列‘ID’,使用merge函数合并
express <- merge(x = exprSet, y = ids, by.x = "ID")#删除探针ID列
express$ID =NULLdim(express) matrix = express
dim(matrix)
#查看多少个基因重复了
table(duplicated(matrix$Gene.Symbol))#把重复的Symbol取平均值
matrix <- aggregate(.~Gene.Symbol, matrix, mean) ##把重复的Symbol取平均值
row.names(matrix) <- matrix$Gene.Symbol #把行名命名为SYMBOLdim(matrix)matrix_na = na.omit(matrix) #去掉缺失值
dim(matrix_na)
matrix_final = matrix_na[matrix_na$Gene.Symbol != "",]
dim(matrix_final)matrix_final <- subset(matrix_final, select = -1) #删除Symbol列(一般是第一列)
dim(matrix_final)
> dim(matrix_final)
[1] 14826 31
小结
原始数据记录有22283条,多个探针对应一个基因采用取平均值处理,一个探针对应多个基因分别进行直接删除操作和保留第一个基因操作, 两种方法最终获得的数据记录分别为12548,14826。
更新版本的代码全文
# 安装并加载GEOquery包
library(GEOquery)# 指定GEO数据集的ID
gse_id <- "GSE1297"# 使用getGEO函数获取数据集的基础信息
gse_info <- getGEO(gse_id, destdir = ".", AnnotGPL = F ,getGPL = F) # Failed to download ./GPL96.soft.gz!# 提取基因表达矩阵
expression_data <- exprs(gse_info[[1]])# 提取注释信息
annotation <- featureData(gse_info[[1]])#查看平台文件列名
colnames(annotation)#打印项目文件列表
dir() # 读取芯片平台文件txt
platform_file <- read.delim("GPL96-57554.txt", header = TRUE, sep = "\t", comment.char = "#")#查看平台文件列名
colnames(platform_file)# 假设芯片平台文件中有两列,一列是探针ID,一列是基因名
#probe_names <- platform_file$ID
#gene_symbols <- platform_file$Gene.Symbol
platform_file_set=platform_file[,c(1,11)]#一个探针对应多个基因名,保留第一个基因名
ids = platform_file_set
library(tidyverse)
test_function <- apply(ids,1,function(x){paste(x[1],str_split(x[2],'///',simplify=T),sep = "...")})
x = tibble(unlist(test_function))colnames(x) <- "ttt"
ids <- separate(x,ttt,c("ID","Gene.Symbol"),sep = "\\...")
dim(ids)#将Matrix格式表达矩阵转换为data.frame格式
exprSet <- data.frame(expression_data)
dim(exprSet)#给表达矩阵新增加一列ID
exprSet$ID <- rownames(exprSet) # 得到表达矩阵,行名为ID,需要转换,新增一列
dim(exprSet)
#矩阵表达文件和平台文件有相同列‘ID’,使用merge函数合并
express <- merge(x = exprSet, y = ids, by.x = "ID")#删除探针ID列
express$ID =NULLdim(express) matrix = express
dim(matrix)
#查看多少个基因重复了
table(duplicated(matrix$Gene.Symbol))#把重复的Symbol取平均值
matrix <- aggregate(.~Gene.Symbol, matrix, mean) ##把重复的Symbol取平均值
row.names(matrix) <- matrix$Gene.Symbol #把行名命名为SYMBOLdim(matrix)matrix_na = na.omit(matrix) #去掉缺失值
dim(matrix_na)matrix_final = matrix_na[matrix_na$Gene.Symbol != "",]
dim(matrix_final)matrix_final <- subset(matrix_final, select = -1) #删除Symbol列(一般是第一列)
dim(matrix_final)
#+ 经过注释、探针名基因名处理、删除基因名为空值、删除缺失值 得到最终 matrix_final
#+==================================================================================
#+========================================================================================已经完成了部分的预处理工作了,在使用数据前还有一系列的质控要做,请看下节数据清洗。
99%的人还看了
相似问题
- 基因预测软件prodigal的使用
- 【佳学基因检测】Node.js中如何读取并调用内容
- 零代码复现-TCGA联合GEO免疫基因结合代谢基因生信套路(一)
- 泡泡玛特首度跨界超跑品牌兰博基尼汽车,以潮流基因探索时空边界
- LeetCode:2003. 每棵子树内缺失的最小基因值(C++)
- 易基因: Nature Biotech:番茄细菌性青枯病的噬菌体联合治疗|国人佳作
- 重测序基因组:Pi核酸多样性计算
- GEO生信数据挖掘(三)芯片探针ID与基因名映射处理
- 8+单基因+细胞凋亡+WGCNA+单细胞+实验验证
- DeepMind 利用无监督学习开发 AlphaMissense,预测 7100 万种基因突变
猜你感兴趣
版权申明
本文"GEO生信数据挖掘(三)芯片探针ID与基因名映射处理":http://eshow365.cn/6-15648-0.html 内容来自互联网,请自行判断内容的正确性。如有侵权请联系我们,立即删除!
- 上一篇: 十个有用的 Vue.js 自定义 Hook
- 下一篇: 发布以太坊测试网络中的第一笔交易